

In a breakthrough that could reshape how we approach cancer drug discovery, researchers have developed a powerful intracellular screening platform that identifies covalent peptide inhibitors capable of irreversibly shutting down oncogenic transcription factors.
A New Way to Target the ‘Undruggables’
Transcription factors, the master regulators of gene expression, have long presented a major challenge in drug development. These proteins typically lack the well-defined pockets that conventional drugs exploit. Instead, they engage in large, flat protein-DNA or protein-protein interactions, making them difficult to inhibit selectively and effectively.
While antibodies are too large to enter cells, small molecules often fall short when it comes to binding these diffuse surfaces. Peptides, however, offer an ideal middle ground: they are large enough to bind extended interfaces with high specificity, yet small and versatile enough to be engineered for intracellular delivery.
One such transcription factor, cJun, plays a central role in tumor progression and resistance to treatment in various cancers. It binds specific DNA motifs to activate genes that promote proliferation and survival. Blocking cJun function has become a promising strategy to curb cancer at its genetic roots.
Moving Beyond Binding: The Transcription Block Survival (TBS) Assay
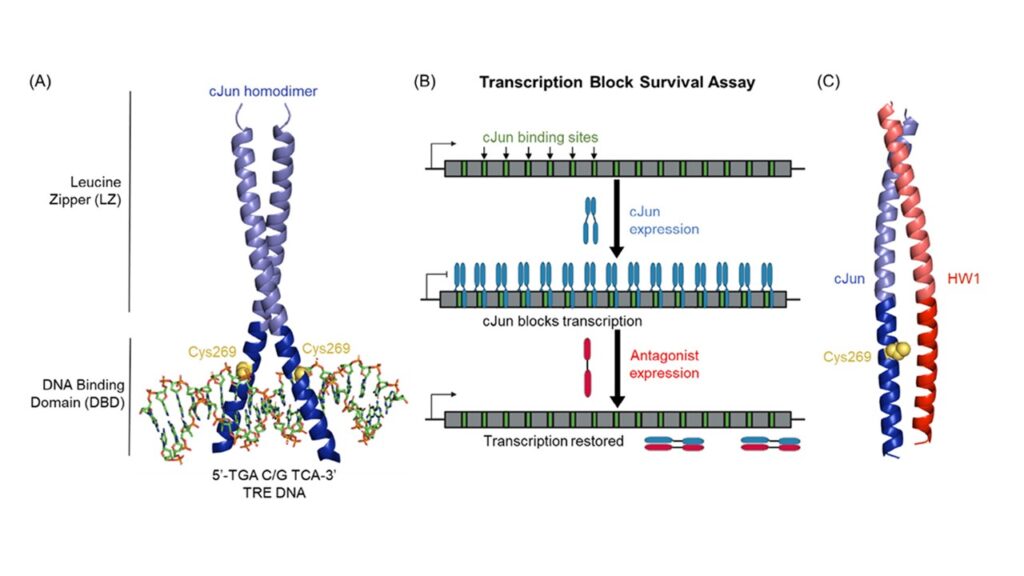
To tackle the challenge, the research team refined a powerful screening method known as the Transcription Block Survival (TBS) assay. Unlike traditional screening systems that rely on detecting whether a molecule binds a target, TBS focuses on whether the molecule can block the protein’s activity inside cells.
The principle is elegantly simple. Engineered bacterial cells require a gene for survival, but that gene contains binding sites for cJun. When cJun is active, it binds and prevents gene expression, stopping cell growth. If a peptide antagonist successfully blocks cJun’s function, the gene is expressed, and the cells grow. This direct genotype-to-phenotype link allows for effective, function-based screening of peptide libraries.
This platform had already led to the discovery of a potent peptide, HW1, which bound cJun reversibly and blocked its DNA binding. But researchers set out to do more: to create a peptide that irreversibly inactivates cJun.
Finding the Right Chemistry: Exploiting a Vulnerable Cysteine
The team focused on a unique cysteine residue—Cys269—within cJun’s DNA-binding domain. This residue, exposed and positioned at the protein-DNA interface, had previously been identified as a redox-sensitive switch capable of turning off cJun’s binding activity.
To exploit this vulnerability, researchers created a massive peptide library of over 131,000 members. Each peptide variant included the possibility of incorporating a cysteine at strategic positions, with the aim of forming a disulfide bond with cJun’s Cys269.
This library was screened in two strains of E. coli: one that promoted disulfide bond formation (oxidizing conditions) and one that did not (reducing conditions). In oxidizing conditions, a standout peptide—named OxW—was selected. It included a cysteine in the ideal position to form a bond with Cys269, effectively shutting down cJun’s activity. In reducing conditions, where disulfide bonds cannot form, the cysteine was not selected, underscoring the importance of this covalent interaction.
From Disulfide to Irreversible: Engineering HW33
The OxW peptide was further refined into HW31, incorporating structural features like a lactam bridge to stabilize its conformation. HW31 demonstrated strong binding and functional inhibition through disulfide bond formation with cJun.
However, disulfide bonds are reversible and sensitive to the cellular environment. To create an irreversible inhibitor, the cysteine in HW31 was replaced with two different chemical groups. The first, dehydroalanine, resulted in peptide HW32. The second, a bis-alkylating electrophile, led to HW33.
HW33 rapidly formed a permanent covalent bond with cJun in vitro, completing the reaction within minutes. Importantly, it did not react with CREB1, a related transcription factor that shares similar cysteine residues. This confirmed that the reaction was highly specific and dependent on the structural interaction between peptide and target.
Getting Inside the Cell: Turning HW33 into a Therapeutic Candidate
To translate these findings into a cellular context, researchers modified HW33 by attaching a cell-penetrating peptide and a nuclear localization signal, forming FAM-HW33-NLSTAT. This enhanced version could efficiently enter human melanoma (SK-MEL-28) cells in culture.
Once inside the cells, the peptide successfully depleted cJun protein levels and significantly reduced cell viability. These effects were observed at concentrations as low as 10 µM and became more pronounced over time, suggesting that the covalent inhibition was both durable and biologically effective.
Western blot analysis revealed that cJun protein levels dropped substantially within 6 hours of treatment and were nearly undetectable after 24 hours. This reduction occurred in a dose-dependent manner. The mechanism is not yet fully understood, but it likely involves accelerated degradation of the modified cJun protein.
Interestingly, the peptide’s activity did not depend on the full nuclear localization. Even when its distribution was largely cytoplasmic, the peptide was effective—possibly by intercepting newly synthesized cJun before it reaches the nucleus.
A Versatile Platform for Future Therapies
Beyond the success of HW33, the broader implication of this study lies in the screening method itself. By using genetically encoded peptide libraries and intracellular screening under oxidizing conditions, researchers can now identify both functional inhibitors and optimal sites for covalent targeting on proteins.
This approach enables rapid discovery of highly selective covalent peptides, even in the absence of high-resolution structural information about the target protein. Moreover, it sidesteps some of the pitfalls of synthetic high-throughput screening by operating in a biological context from the start.
Because the system favors peptides that are stable, selective, and functional inside cells, the hits it produces are inherently more likely to succeed in therapeutic development.
Looking Ahead
While the study focused on cJun as a proof of concept, the potential applications of this platform extend much further. Many transcription factors and other disease-relevant proteins contain cysteines that may be similarly exploited. With customized libraries and improved delivery mechanisms, the TBS screening approach could lead to a new class of peptide-based drugs capable of tackling previously intractable targets.
The development of HW33 shows that it is possible to design peptides that not only bind with high affinity and specificity but also form irreversible covalent bonds that amplify their functional effect. This work marks a promising step forward in the evolution of targeted cancer therapies.
Paolo Rega
Leave a Reply